Pulmonale Hypertonie
(auch als padBook erhältlich)
Definitionen
Eine pulmonale Hypertonie liegt vor, wenn der pulmonalarterielle Mitteldruck ≥25 mm Hg beträgt (Normalwert: 20 mm Hg). Der Grauwert von 20 - 25 mm Hg ist bislang nicht eindeutig definiert.
In früheren Zeiten gab es zusätzlich das Kriterium eines PA-Mitteldruckanstiegs von >30 mm Hg unter Belastung. Dieses Kriterium wurde allerdings wieder verlassen, weil der Mitteldruck bei verschiedenen gesunden (vor allem bei älteren) Menschen auf weit höhere Werte als 30 mm Hg ansteigen kann, ohne daß eine bedeutsame Herz-Kreislauf-Erkrankung vorliegen würde. Daher kann man derzeit keinen Grenzwert angeben, von dem an ein PA-Mitteldruck als pathologisch zu betrachten ist.
Klassifikation
Man klassifiziert die pulmonalen Hypertonien nach verschiedenen Gesichtspunkten:
Ätiologische Klassifikation
Die Klassifikation der pulmonalen Hypertonie erfolgt gemäß der WHO folgendermaßen:
Nachfolgend finden Sie einige Erläuterungen zu den verschiedenen Erkrankungen:
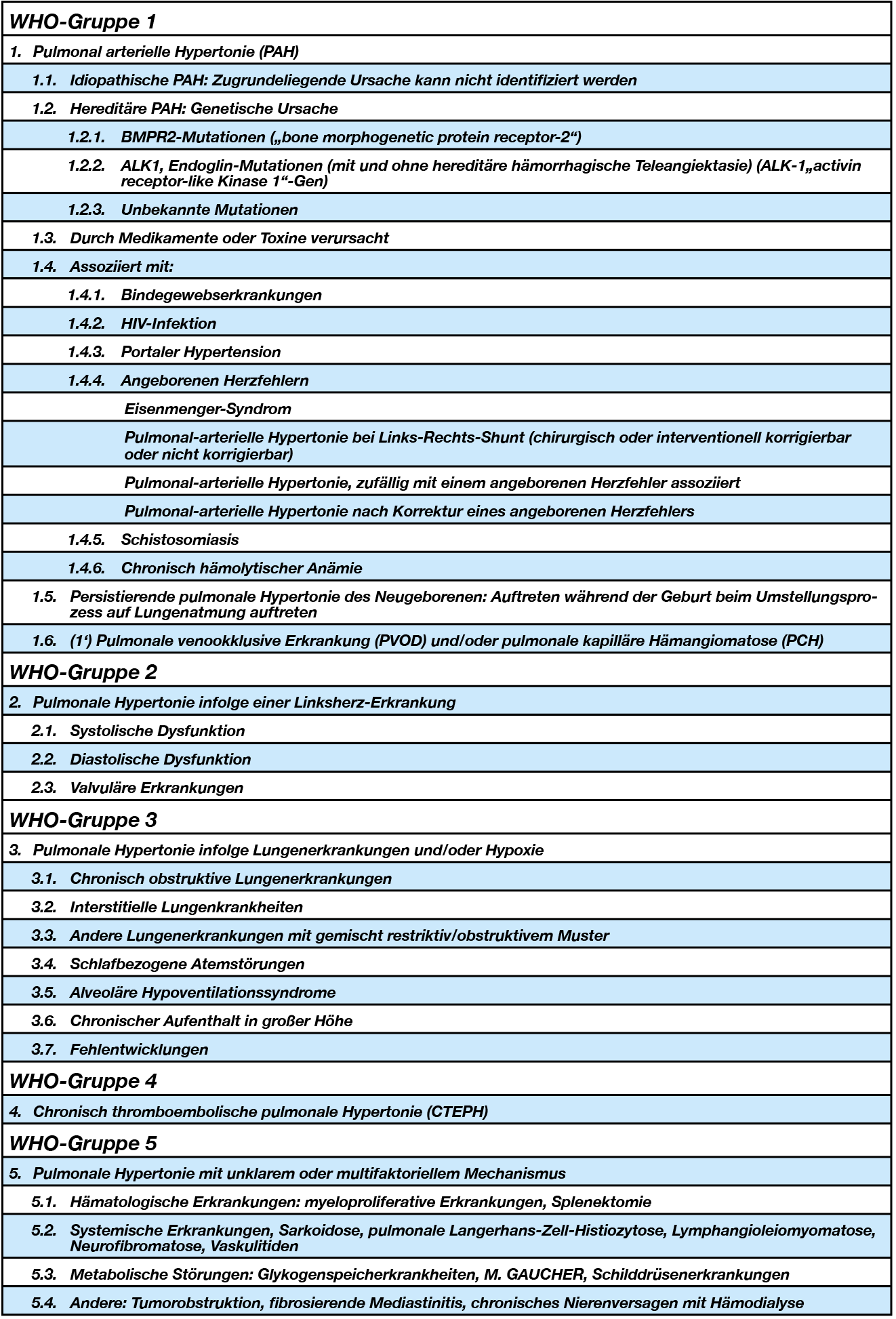
WHO-Gruppe 1
Gruppe 1 umfaßt Erkrankungen mit ähnlichen histopathologischen Veränderungen:
- Verdickung der Tunica media der Gefäßwand und Dilatation der präazinären Pulmonalarterien (Abb. 1)
- zelluläre Intimaproliferation
- Ausbildung von lokalen Thrombosen
- Intimaproliferation und -fibrose und
- plexiforme Läsionen, die als Umgehungskreisläufe (Plexus) durch Vasa vasorum bei fibrotisch verschlossenen Pulmonalarterien ausgebildet werden (Abb. 2).
 |

|
| Abb. 1 |
Abb. 2 |
Alle pathologischen Veränderungen können gleichzeitig vorkommen. Die histologische Bewertung erfolgt daher nicht in einer Gradeinteilung, sondern deskriptiv qualitativ.
Weil die Lungenstrombahn bei den Erkrankungen dieser Gruppe primär betroffen ist spricht man auch von pulmonal-arterieller Hypertonie.
Pulmonal arterielle Hypertonie (PAH)
Idiopathische PAH: Zugrundeliegende Ursache kann nicht identifiziert werden
Hereditäre PAH: Genetische Ursache
- BMPR2-Mutationen („bone morphogenetic protein receptor-2“)
- ALK1, Endoglin-Mutationen (mit und ohne hereditäre hämorrhagische Teleangiektasie) (ALK-1„activin receptor-like Kinase 1“-Gen)
- Unbekannte Mutationen
Durch Medikamente oder Toxine verursacht
Assoziiert mit:
- Bindegewebserkrankungen
- HIV-Infektion
- Portaler Hypertension
- Angeborenen Herzfehlern:
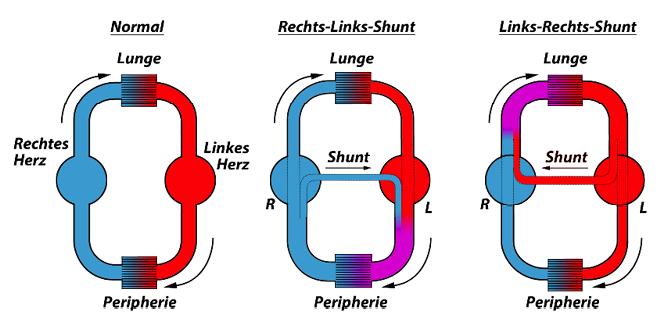
Eisenmenger-Syndrom:
Herz /Gefäßanomalien mit initialem Links-Rechts-Shunt, bei denen im Verlauf der Erkrankung der Lungengefäßwiderstand stark ansteigt und es konsekutiv zunächst zu einem bidirektionalen Shunt oder zur vollständigen Shuntumkehr („Rechts-Links-Shunt“) kommt.
Klinisch ist das Syndrom gekennzeichnet durch eine Zyanose, eine sekundäre Erythrozytose und zyanosebedingte Multiorganbeteiligungen.
Pulmonal-arterielle Hypertonie bei Links-Rechts-Shunt (chirurgisch oder interventionell korrigierbar oder nicht korrigierbar):
Mittelgroße bis große Defekte mit gering- bis mittelgradigem Links-Rechts-Shunt, aber ohne Zyanose unter Ruhebedingungen
Pulmonal-arterielle Hypertonie, zufällig mit einem angeborenen Herzfehler assoziiert
Der angeborene Herzfehler ist nicht Ursache der pulmonal-arteriellen Hypertonie..
Achtung: Der gemessene Diameter eines Shuntostiums kennzeichnet nicht immer die hämodynamische Relevanz des Vitiums. Zur exakteren Beurteilung der Shunt-Hämodynamik müssen Druckgradienten, Shuntgröße und -richtung, sowie das Verhältnis von Lungenblut- und Systemblutfluss [Qp/Qs] berücksichtigt werden.
Es besteht ein deutlich überhöhter Lungengefäßwiderstand in Gegenwart kleiner angeborener Defekte, die nicht primär für die Entwicklung des überhöhten Widerstandes verantwortlich sind.
Das klinische Bild ähnelt stark einer idiopathischen PAH. Ein Defektverschluss ist kontraindiziert.
Pulmonal-arterielle Hypertonie nach Korrektur eines angeborenen Herzfehlers:
Nach operativer Korrektur des Fehlers persistierende oder innerhalb von Monaten oder Jahren wiederkehrende pulmonal-arterielle Hypertonie ohne hämodynamisch relevante Re-/Restshunts
- Schistosomiasis
- Chronisch hämolytischer Anämie
- Persistierende pulmonale Hypertonie des Neugeborenen: Auftreten während der Geburt beim Umstellungsprozess auf Lungenatmung auftreten
- (1‘) Pulmonale venookklusive Erkrankung (PVOD) und/oder pulmonale kapilläre Hämangiomatose (PCH)
Im Gegensatz dazu liegen bei den Krankheiten der Gruppen 2 - 5 andere Ursachen vor, die über spezielle (jedoch z.T. noch unbekannte Mechanismen zur pulmonalen Hypertonie führen. Diese können zwar auch zu strukturellen Veränderungen der präkapillären und kapillären Strombahn führen, jedoch sind diese Veränderungen sekundärer Natur.
WHO-Gruppe 2
- Pulmonale Hypertonie infolge einer Linksherz-Erkrankung
- Systolische Dysfunktion
- Diastolische Dysfunktion
- Valvuläre Erkrankungen
WHO-Gruppe 3
- Pulmonale Hypertonie infolge Lungenerkrankungen und/oder Hypoxie
- Chronisch obstruktive Lungenerkrankungen
- Interstitielle Lungenkrankheiten
- Andere Lungenerkrankungen mit gemischt restriktiv/obstruktivem Muster
- Schlafbezogene Atemstörungen
- Alveoläre Hypoventilationssyndrome
- Chronischer Aufenthalt in großer Höhe
- Fehlentwicklungen
WHO-Gruppe 4
- Chronisch thromboembolische pulmonale Hypertonie (CTEPH)
WHO-Gruppe 5
Pulmonale Hypertonie mit unklarem oder multifaktoriellem Mechanismus
- Hämatologische Erkrankungen: myeloproliferative Erkrankungen, Splenektomie
- Systemische Erkrankungen, Sarkoidose, pulmonale Langerhans-Zell-Histiozytose, Lymphangioleiomyomatose, Neurofibromatose, Vaskulitiden
- Metabolische Störungen: Glykogenspeicherkrankheiten, M. GAUCHER, Schilddrüsenerkrankungen
- Andere: Tumorobstruktion, fibrosierende Mediastinitis, chronisches Nierenversagen mit Hämodialyse
Von allen genannten Erkrankungen ist eine pulmonale Hypertonie am häufigsten die Folge:
- chronisch-obstruktiven Lungenerkrankung (COPD). Hierbei, z.B. bei der COPD kommt es zu Veränderungen des Lungengewebes, die die Lungenfunktion einschränken. Dies wiederum hat direkten Einfluss auf den Lungenkreislauf und kann hier zum Bluthochdruck führen. Chronischer Sauerstoffmangel kann generell eine pulmonale Hypertonie verursachen (siehe VON EULER-LILLJESTRAND-Reflex). Diese Unterform ist relativ häufig.
Abb. 3
-
Thromboembolien. Es wird davon ausgegangen, daß dieser Form der pulmonalen Hypertonie häufig eine Lungenembolie vorausgegangen ist (Abb. 3). Bei dieser relativ seltenen Form löst sich der Thrombus nicht selbstständig auf, verwächst mit der Gefäßwand und bekommt eine faserige Konsistenz. Der Blutfluss wird behindert und der Blutdruck erhöht sich. Hier kann ein chirurgischer Eingriff (pulmonale Endarteriektomie) heilend sein. Bei der Operation wird der eingewachsene Gefäßverschluss aus dem betroffenen Lungenblutgefäß entfernt. Diese Operation ist aber nicht bei allen Patienten möglich.
bei erhöhtem linksventrikulärem Füllungsdruck
bei angeborenen Shuntvitien mit pulmonaler Hyperzirkulation, sowie
interstitieller Lungenerkrankungen, die mit einer perivaskulären Entzündung und einem Verlust an alveolokapillären Funktionseinheiten einhergehen.
Funktionelle Klassifikation
Eine Einteilung, die für die Beschreibung der klinischen Schwere der Erkrankung benutzt wird, die aber auch und vor allem für die Auswahl der medikamentösen Therapie von Bedeutung ist stellt die WHO-Funktionsklassifizierung dar. Sie entspricht der in der Kardiologie üblichen NYHA-Klassifizierung:
- Klasse 1
Patienten mit pulmonaler Hypertonie ohne Einschränkung der körperlichen Aktivität. Normale körperliche Belastungen führen nicht zu vermehrter Dyspnoe oder Müdigkeit, thorakalen Schmerzen oder Schwächeanfällen
- Klasse 2
Patienten mit pulmonaler Hypertonie mit einer leichten Einschränkung der körperlichen Aktivität. Keine Beschwerden in Ruhe. Normale körperliche Aktivität führt zu vermehrter Dyspnoe oder Müdigkeit, thorakalen Schmerzen oder Schwächeanfällen
- Klasse 3
Patienten mit pulmonaler Hypertonie mit deutlicher Einschränkung der körperlichen Aktivität. Keine Beschwerden in Ruhe. Bereits leichtere als normale Belastungen führen zu Dyspnoe oder Müdigkeit, thorakalen Schmerzen oder Schwächeanfällen
- Klasse 4
Patienten mit pulmonaler Hypertonie die keinerlei körperliche Belastung ohne Beschwerden aus- führen können. Zeichen der manifesten Rechtsherzinsuffizienz. Dyspnoe und/oder Müdigkeit können bereits in Ruhe vorhanden sein. Bei geringster Aktivität werden die Beschwerden verstärkt
Pathophysiologische Klassifikation
Aus pathophysiologischer Sicht unterscheidet man die präkapilläre von der postkapillären pulmonalen Hypertonie, sowie den seltenen Sonderfall einer kapillären pulmonalen Hypertonie (z.B. bei der pulmonalen kapillären Hämangiomatose).
| Definition |
Charakteristika |
| Präkapilläre PH |
PAPm ≥25 mm Hg |
| |
PC ≤15 mm Hg |
| |
HZV normal oder erniedrigt |
| Postkapilläre PH |
PAPm ≥25 mm Hg |
| |
PC >15 mm Hg |
| |
HZV normal oder erniedrigt |
| |
TPG ≤12 mm Hg |
| Komb. prä- und postkapilläre PH |
TPG >12 mm Hg |
| PH pulmonale Hypertonie, PAPm mittlerer pulmonal-arterieller Druck, PC„pulmonary-capillary wedge pressure“ bzw. pulmonal-kapillärer Verschlussdruck, TPG transpulmonaler Druckgradient (PAPm-PCWP). |
Häufigkeit
Unter Einbeziehung aller Varianten der pulmonalen Hypertonie ist Schätzungen zufolge etwa 1% aller Menschen weltweit betroffen. Die zahlenmäßig größte Gruppe bilden vermutlich die Betroffenen, die an einer strukturellen oder funktionellen Erkrankung des linken Herzens leiden.
Die exakte Häufigkeit der idiopathischen und hereditären pulmonalen Hypertonie bei Kindern und Erwachsenen ist nicht bekannt. Schätzungen europäischer Studien gehen von einer Inzidenz von 0,48/ 1 Mio. Kinder pro Jahr aus, die Prävalenz wird auf etwa 2-2,2 Fälle/1 Mio. Kinder geschätzt.
Wie bei Erwachsenen mit idiopathischen und hereditären ist das weibliche Geschlecht in einem Verhältnis von ca. 1,7:1 häufiger betroffen.
Als Verhältnis von Kindern, die an Herzfehler-assoziierter pulmonaler Hypertonie erkranken zu Kindern mit idiopathischen und hereditären Lungenhochdrücken wird etwa 1 : 0,9 angegeben.
Risikofaktoren
Es gibt Risikokonstellationen, die das Auftreten einer pulmonalen Hypertonie begünstigen. Die hierdurch hervorgerufene Hypertonie läßt sich klinisch und histologisch nicht von der primären pulmonalen Hypertonie unterscheiden.
Zu solchen Risikofaktoren gehören die familiären Vorgeschichte mit primär pulmonal-arterieller Hypertonie, die eine genetische Prädisposition mit erhöhtem Risiko für das Auftreten der Erkrankung bedeutet.
Dieses „genetische Risiko“ scheint auch eine Rolle dabei zu spielen, daß sich bei bestimmten Ereignissen oder dem Auftreten anderer Erkrankung eine pulmonale Hypertonie entwickelt:
- HIV-Infektion
- Autoimmunerkrankungen
- Chronisch obstruktive Lungenerkrankung (COPD)
- Lungenembolie
- Linksherzerkrankung
- Schistosomiasis (Bilharziose)
- Leberzirrhose (erhöhtes Risiko für eine portale pulmonale Hypertonie, ohne daß der ätiologische Zusammenhang bisher verstanden wird)
- bestimmte Medikamente:
- Zusammenhang zu pulmonaler Hypertonie gesichert:
- Amiodarone
- Fenfluramine
- Dexfenfluramine
- toxisches Rapsöl
- Benfluorex (Antidiabeticum)
- Serotonin-Wiederaufnahmehemmer (Antidepressivum, bei Einnahme während der Schwangerschaft pulmonale Hypertonie beim Neugeborenen)
- Zusammenhang wahrscheinlich:
- Amphetamine
- Metamphetamine
- l-Tryptophan
- Dasatinib (Chemotherapeutikum zur Behandlung bestimmter maligner Erkrankung (chronisch melodische und akute lymphatische Leukämie)
- Zusammenhang möglich:
- Kokain
- Phenylpropanolamin (stimulierender und appetithemmender Wirkstoff aus der Gruppe der Sympathomimetika zur Behandlung von Übergewicht und Erkältungserkrankungen)
- Johanniskraut
- Amphetamin-ähnliche Medikamente (z.B. Ephedrin, Cathinon, Phentermin, Mephentermine, Bupropion (Antidepressivum, zur Raucherentwöhnung Anorecticum), Methoxyphenamin, Selegilin (Anti-Parkinson-Mittel), Amfepramon (Behandlung der Adipositas), Pyrovaleron (Stimulans bei chronischer Müdigkeit, Adipositas, Lethargie), Ecstasy, 2,5-Dimethoxy-4-methylamphetamin (DOM).
- Interferon alpha und beta
- Einige alkylierende Chemotherapeutika (z.B. Cyclophosphamid, Myotomyzin C) (venookklusive PA)
Auch im Zusammenhang mit Lungenkrankheiten wie COPD scheint diese genetische Prädisposition eine Rolle zu spielen.
Pathogenese
Angesichts der Vielzahl der Erkrankungen, die zur pulmonalen Hypertonie führen können gibt es natürlich auch eine Vielzahl pathogenetischer Faktoren. Einige dieser Mechanismen sind bekannt, viele jedoch bis heute unbekannt.
Es gibt „lediglich“ 3 Prinzipien, über die man die pulmonale Druckerhöhung erklären kann:
- Gefäßobstruktion
Bei Einengungen (Obstruktionen) der Lungengefäße kommt das OHM´sche Gesetz zur Wirkung:
R (Widerstand) = U (Spannung) / I (Fluss)
oder anders herum
U = R * I (Druckgradient = Gefäßwiderstand * Blutfluß).
Dies bedeutet logischerweise, daß die Zunahme des Gefäßwiderstandes bei gleich bleibendem Blutfluß zu einer Erhöhung des Druckgradienten über der Lunge führen. Und wenn der pulmonal-venöse Druck niedrig bleibt muß folglich der pulmonalarterielle Druck ansteigen.
- Hypoxie
Grundsätzlich weist das pulmonale Gefäßbett gegenüber dem Systemkreislauf die folgenden grundsätzlichen Unterschiede auf:
Pulmonale Arteriolen reagieren auf eine Hypoxie mit einer Vasokonstriktion (EULER-LILJESTRAND-Reflex), während im Systemkreislauf eine Vasodilatation erfolgt. Die Obstruktion der Arteriolen führt dann (siehe oben unter „Gefäßobstruktion“) zur pulmonal-arteriellen Druckerhöhung.
- Intensität des Blutflusses durch die Lungengefäße
Bei steigendem Blutfluß erfolgt im Lungenkreislauf eine aktive Vasodilatation und eine Rekrutierung apikaler, normalerweise wenig perfundierter Lungenabschnitte, sodaß der Druck in der A. pulmonalis weitgehend konstant gehalten wird.
Verantwortlich für diesen Autoregulationsmechanismen sind endothelabhängige Vasodilatatoren wie Stickstoffmonoxid (NO) und Prostazyklin (PGI2).
Daher scheint die Ursache für die Entwicklung einer pulmonalen Hypertonie eine Störung der Autoregulation im Lungenkreislauf zu sein, bei der es zu einem Ungleichgewicht zwischen Vasodilatatoren und Vasokonstriktoren zugunsten der letzteren kommt.
Als vasokonstriktorische Mechanismen wirken
Zuordnung der einzelnen Erkrankungen
Nachfolgend habe ich nun versucht, die verschiedenen Erkrankungen den 3 Grundtypen der pulmonalen Hypertonie zuzuordnen und dabei die jeweilige Pathogenetik zu erwähnen.
Präkapilläre pulmonale Hypertonie
Es handelt sich um eine Gefäßobstruktion im Bereich der Arteriolen der arteriellen Lungenstrombahn. Die Folge ist ein Druckanstieg in den präkapillär gelegenen arteriellen Gefäßen.
Verursacht werden kann sie durch
- chronisch-obstruktive Lungenerkrankungen:
- Bronchopneumonie:
- Paravaskuläre Entzündung mit „externer Kompression“ und damit Obstruktion der Lungengefäße
- Hypoxie mit EULER-LILJESTRAND-Reflex
- interstitielle Lungenerkrankungen, Lungenfibrose:
- Gefäßrarefizierung und hierdurch Widerstandserhöhung der arteriellen Strombahn
- direkte oder indirekte Beteiligung der Alveolen und hierdurch bedingt Hypoxie mit EULER-LILJESTRAND-Reflex
- Kollagenosen:
Können Atemmuskulatur, Pleura, Lungenparenchym (Pneumonitis oder Lungenfibrose) und das Gefäßsystem betreffen.
Die pathogenetischen Mechanismen für die pulmonal-arterielle Druckerhöhung sind somit in Abhängigkeit der Auswirkungen der Kollagens auf die genannten Strukturen:
- Gefäßrarefizierung und hierdurch Widerstandserhöhung der arteriellen Strombahn
- obstruktive Veränderungen der arteriellen Lungengefäße bei direkter Beteiligung dieser Gefäße an der Kollagenose
- Hypoxie mit EULER-LILJESTRAND-Reflex bei Beteiligung der Atemmuskulatur
- direkte oder indirekte Beteiligung der Alveolen und hierdurch bedingt Hypoxie mit VON EULER-LILJESTRAND-Reflex
- Vasculitiden:
- obstruktive Veränderungen der arteriellen Lungengefäße
- alveoläre Hypoventilation, Schlafapnoe-Syndrome:
- Lungenembolie:
- obstruktive Veränderungen der arteriellen Lungengefäße
- Extrakardiale Hypoxie:
- Angeborene Herzfehler:
- Bei pulmonalen Druckerhöhungen ohne Shunt liegt in der Regel entweder eine genetisch bedingte primär pulmonal-arterielle Hypertonie vor oder es handelt sich um die Auswirkung von bestimmten Medikamenten, die die Mutter während der Schwangerschaft eingenommen hat.
Hämodynamisch gesehen kann die rechtsventrikuläre Funktion die progrediente Erhöhung des pulmonalen Gefäßwiderstandes im Verlauf ggf. nicht mehr kompensieren mit der Folge einer führenden Rechts- und schließlich globalen Herzinsuffizienz. Dies betrifft im Grundsatz alle anderen Fälle mit pulmonaler Hypertonie auch.
links: Abb. 4
- Bei pulmonalen Hypertonie bei Patienten mit Shuntverbindung auf Herz- oder Gefäßebene ist derjenige Mechanismus verantwortlich, der weiter oben unter „Intensität des Blutflusses durch die Lungengefäße“ geschrieben wurde (Abb. 4).
Bei denjenigen Patienten, bei denen ein hoher pulmonal-arterieller Widerstand besteht und bei denen der Shunt als Überdruckventil fungiert kann dieser Shunt sogar als einen begünstigenden Faktor für das Überleben darstellen.
Zusätzlich haben diese Patienten bei der meist schon von Geburt an bestehender pulmonalen Druckerhöhung eine günstigere Langzeitprognose als Patienten mit anderen angeborenen pulmonal-arteriellen Druckerhöhungen, weil der rechte Ventrikel wahrscheinlich bereits intrauterin besser adaptiert ist.
Bei der fortgeschrittensten Form der Shunt-assoziierten pulmonalen Hypertonie, dem EISENMENGER-Syndrom, entwickeln sich durch die chronische zentrale Zyanose und Hypoxämie zusätzlich sekundäre, komplexe Multiorganfunktionsstörungen.
Kapilläre pulmonale Hypertonie
Es handelt sich um eine Gefäßobstruktion im Bereich der Lungenkapillaren. Die Folge ist auch hier ein Druckanstieg in den präkapillär gelegenen arteriellen Gefäßen.
- Primäre pulmonal-arterielle Hypertonien:
Die Erkrankung tritt überwiegend sporadisch, seltener postpartal und in circa 6 % der Fälle hereditär auf, wobei ein autosomal-dominanter Erbgang mit stark variabler Penetranz vorliegt. Es sind verschiedene Mutationen bekannt.
Pathophysiologisch kommt es zu einer Proliferation und Migration von glatten Muskelzellen und eine abnorme Apoptose. Dadurch vergrößern sich Zellen, die in der Wand der kleinen Lungengefäße sitzen (siehe Abb. 1) (z.B. bei der pulmonalen kapillären Hämangiomatose), was zu strukturellen Veränderungen der Lungenkapillaren führt. Dies wiederum führt zu einer Verdickung der Gefäßwände und verschmälert das Lumen der Kapillaren, was den Blutdurchfluß erschwert und den präkapillären Druck steigert.
In vielen Fällen sieht man histologisch zudem sog. plexiforme Läsionen (siehe Abb. 2), die als Rekanalisation obliterierter Gefäßabschnitte interpretiert werden.
Nach histologischen Untersuchungen stehen neben der funktionellen und strukturellen Vasokonstriktion vor allem ein umfassendes Gefäßremodelling und das Auftreten von In-situ-Thrombosen im Mittelpunkt der Pathogenese.
Trotz unterschiedlicher Mutationen ist das histologische Bild der primären pulmonal-arteriellen Hypertonien in fortgeschrittenen Fällen nahezu identisch, sodaß ein gemeinsamer pathogenetischer Mechanismus angenommen werden kann.
Bei der primären pulmonal-arteriellen Hypertonie findet man ebenso wie bei der präkapillaren pulmonalen Hypertonie eines Erhöhung des pulmonal-arteriellen Mitteldrucks auf >25 mm Hg bei normalem PC-Druck.
- Schwere chronisch-obstruktive Lungenkrankheit (COPD):
- Bronchopneumopathie mit Alveolarüberblähung:
- Langzeit-PEEP-Beatmung (Erhöhung des endexspiratorischer Drucks):
- „externe Kompression“ und damit Obstruktion der Lungengefäße
Postkapilläre pulmonale Hypertonie
- Erhöhter LV-Füllungsdruck:
- Rückstau des erhöhten enddiastolischen LV-Drucks in die Lungenvenen und hierdurch „Abflußbehinderung“ des Blutes in den linken Vorhof und linken Ventrikel mit Druckerhöhung in den Lungenvenen und im weiteren Verlauf bis in die präkapilläre Lungenstrombahn
- Erhöhter Druck im linken Vorhof:
- Siehe „Erhöhter LV-Füllungsdruck“
- Einengung/Obliteration der Lungenvenen, zum Beispiel venookklusive Lungenerkrankung
- Siehe „Erhöhter LV-Füllungsdruck“
Andere Ursachen
Über die zuvor genannten Formen hinaus gibt es noch weitere Ursachen für pulmonale Hypertonie. So können ganz unterschiedliche systemische Erkrankungen, wie z.B. Schilddrüsenerkrankungen oder ein chronischer Nierenausfall, einen Hochdruck im Lungenkreislauf hervorrufen.
Sie bleibt die Methode der Wahl zum Ausschluss einer chronischen thromboembolischen pulmonalen Hypertonie.
Sensitivität und Spezifität betragen 90 – 100 bzw. 94 – 100%, sodaß ein normaler Befund eine thromboembolische Ursache mit hinreichender Sicherheit ausschließen kann. In Zweifelsfällen sollte man eine Lungenembolie mittels Thorax-CT ausschließen bzw. bestätigen.
Die Untersuchung sollte bei jeder schweren pulmonalen Hypertonie Bestandteil der initialen Diagnostik sein (Abb. 7).
CT des Thorax
Ein CT ist fester Bestandteil der Diagnostik von interstitiellen Lungenerkrankungen und Lungenemphysem.
Zudem kann es mit fleckförmigen Milchglastrübungen, verdickten intra- und interlobulären Septen, mediastinaler Lymphadenopathie und Pleuraergüssen Hinweise auf das mögliche Vorliegen einer pulmonalen venookklusiven Erkrankung geben.
1.png) |
.png)
|
Abb. 8
CT bei chronisch thromboembolischer pulmonaler Hypertonie
„Mosaik-Perfusion“ mit segmentaler verstärkter und verminderter Perfusion |
Abb. 9
CT bei chronisch thromboembolischer pulmonaler Hypertonie
Links: Verengte Mittellappenarterie rechts (weißer Pfeil). Rekanalisierter Thrombus in der linken Mittellappenarterie (grüner Pfeil)
Rechts: Starke Verengung der rechts deszendierenden Pulmonalarterie durch einen organisierten Thrombus (solider weißer Pfeil) und membranöse Struktur als Rest eines Thrombus (offener weißer Pfeil) in der linken unteren Segmentarterie |
Das Thorax-CT mit intravenöser Gabe von Kontrastmittel dient neben der Pulmonalisangiographie der weiteren Abklärung einer chronischen thromboembolischen pulmonalen Hypertonie, auch im Hinblick auf eine mögliche Operabilität (Abb. 8 und 9).
Die CT-Diagnostik wird daher als fester Bestandteil der Diagnostik angesehen.
Laboruntersuchungen
Zu den wichtigsten gezielten Laboruntersuchungen bei der Abklärung einer ätiologisch unklaren pulmonalen Hypertonie gehören
- antinukleäre Antikörper inklusive Scl-70
- Centromer- und U1-RNP-Antikörper
- HIV- und Hepatitis-B/C-Serologie und
- eine TSH- Bestimmung (Prävalenz von Thyreopathien bei idiopathischer pulmonalen Hypertonie bis zu 20%).
Die Bestimmung der BNP- bzw. proBNP-Werte kann im Rahmen der initialen Diagnostik und für Verlaufsuntersuchungen hilfreich sein. Jedoch sind BNP/proBNP-Erhöhungen nicht spezifisch für eine Rechtsherzinsuffizienz oder pulmonale Hypertonie.
Rechtsherzkatheter und Vasoreagibilitätstest
Für die Diagnose der pulmonalen Hypertonie selbst stellt die Rechtsherzkatheterisierung mittels Swan-Ganz-Katheter den Goldstandard dar (siehe hierzu auch die Kapitel 12 über die „Rechtsherzkatheteruntersuchung“ und Kapitel 10 über „Messungen“ im „Corobuch“). Sie ist erforderlich, um die Diagnose einer pulmonalen Hypertonie zu bestätigen, ihre Ätiologie weiter abzuklären und den Schweregrad einzuschätzen.
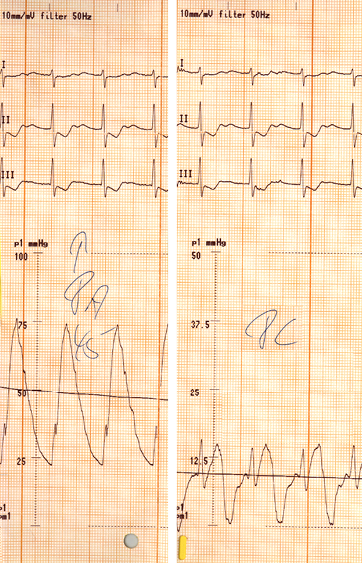
Links: Abb. 10
Druckmessung mit Rechtsherzkatheter bei pulmonaler Hypertonie
Links deutlich erhöhter PA-, rechts niedrig-normaler PC-Druck
Die Untersuchung erlaubt die direkte Messung der Druckverhältnisse im rechten Herzen, im Lungenkreislauf sowie mittels des pulmonalkapillären Verschlußdruckes (PC) auch die Bestimmung des linksventrikulären Füllungsdrucks beziehungsweise des linksatrialen Druckes. Auf diese Weise gelingt auch die Unterscheidung zwischen prä- und postkapillärer pulmonaler Hypertonie (Abb. 10).
Die folgenden Parameter müssen obligat bestimmt werden:
- rechtsatrialer Druck
- pulmonal-arterieller Druck (pPA)
- pulmonal-kapillärer Verschlussdruck (pPC)
- Herzzeitvolumen (Thermodilution oder Fick-Prinzip mit gemessener O2-Aufnahme (tabellarische O2-Aufnahme nicht hinreichend zuverlässig)) und
- gemischt-venöse Sauerstoffsättigung.
Die zentral wichtige Berechnung des totalen Lungengefäßwiderstandes (PVR) erfolgt nach der Gleichung:
PVR [dyn x s x cm-5] = (80 x [PAP - PC]) / HZV
(PAP = mittlerer Pulmonalisdruck, PC = mittlerer PC-Druck)
Eine Vasoreagibilitätstestung dient einzig der Frage, ob Patienten mit Kalziumantagonisten behandelt werden können. Diese Testung wird nur bei Patienten mit pulmonal-arterieller Hypertonie durchgeführt und gilt bei neu diagnostizierter Erkrankung als obligatorisch.
Als Testsubstanzen werden in Deutschland überwiegend inhalatives Stickstoffmonoxid (NO) oder inhalatives Iloprost verwendet.
- NO wird üblicherweise in einer Dosierung von 20 ppm verabreicht, und die Hämodynamik kann bereits nach 5 min durchgehender Inhalation gemessen werden.
- Iloprost wird zumeist in der Dosis von 5 μg inhalativ über 5 min verabreicht, gefolgt von einer Bestimmung der hämodynamischen Parameter nach weiteren 5–10 min.
Kriterien für ein positives Ansprechen (Responder) sind ein Abfall des PA-Mitteldrucks um >10 mm Hg vom Ausgangswert auf <40 mm Hg bei normalem Herzzeitvolumen.
Pulmonalisangiographie
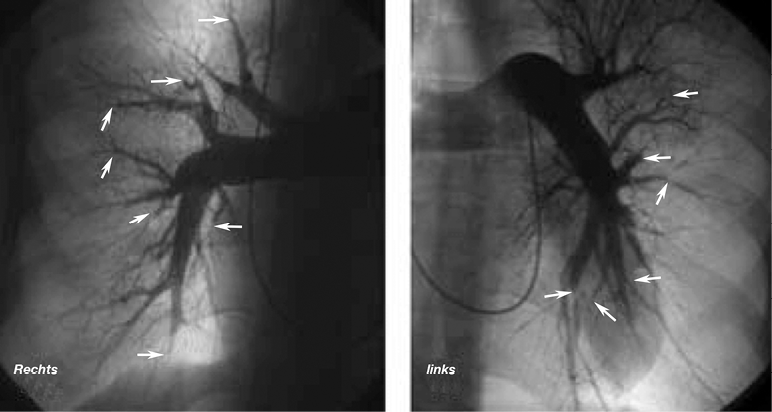
Links: Abb. 11
Pulmonalis-Angio bei Patienten mit chronisch thromboembolischer pulmonaler Hypertonie
In zahlreichen Segment- und Subsegment-Arterien erkennt man Gefäßabbrüche, Kalibersprünge und Stenosen (Pfeile)
In vereinzelten Fällen führt man zusätzlich eine Pulmonalisangiographie durch (Abb. 11), jedoch ist dieses Verfahren nicht ohne Risiken und kann heute mit derselben Treffsicherheit auch durch eine CT- oder MRT-Untersuchung ersetzt werden.
Alle invasiven Untersuchung sollten grundsätzlich nur in Zentren durchgeführt werden, die umfangreiche Erfahrungen in der Diagnostik und Therapie der pulmonalen Hypertonie besitzen. Dies gilt ebenso für CT- oder MRT-Untersuchungen.
In erfahrenen Zentren ist die Komplikationsrate der Untersuchung niedrig (Morbidität 1,1%, Mortalität 0,055%) und die diagnostische Sicherheit für bildgebende Verfahren optimal.
Magnetresonanz-Tomographie (MRT)
Die Untersuchungsmethode spielt bei der Diagnose der pulmonalen Hypertonie keine Rolle. Sie kann aber erforderlich sein, wenn das Vorliegen eines angeborenen Herzfehlers vermutet oder bekannt ist, um die Art des Vitium zu definieren.
Diagnostisches Vorgehen
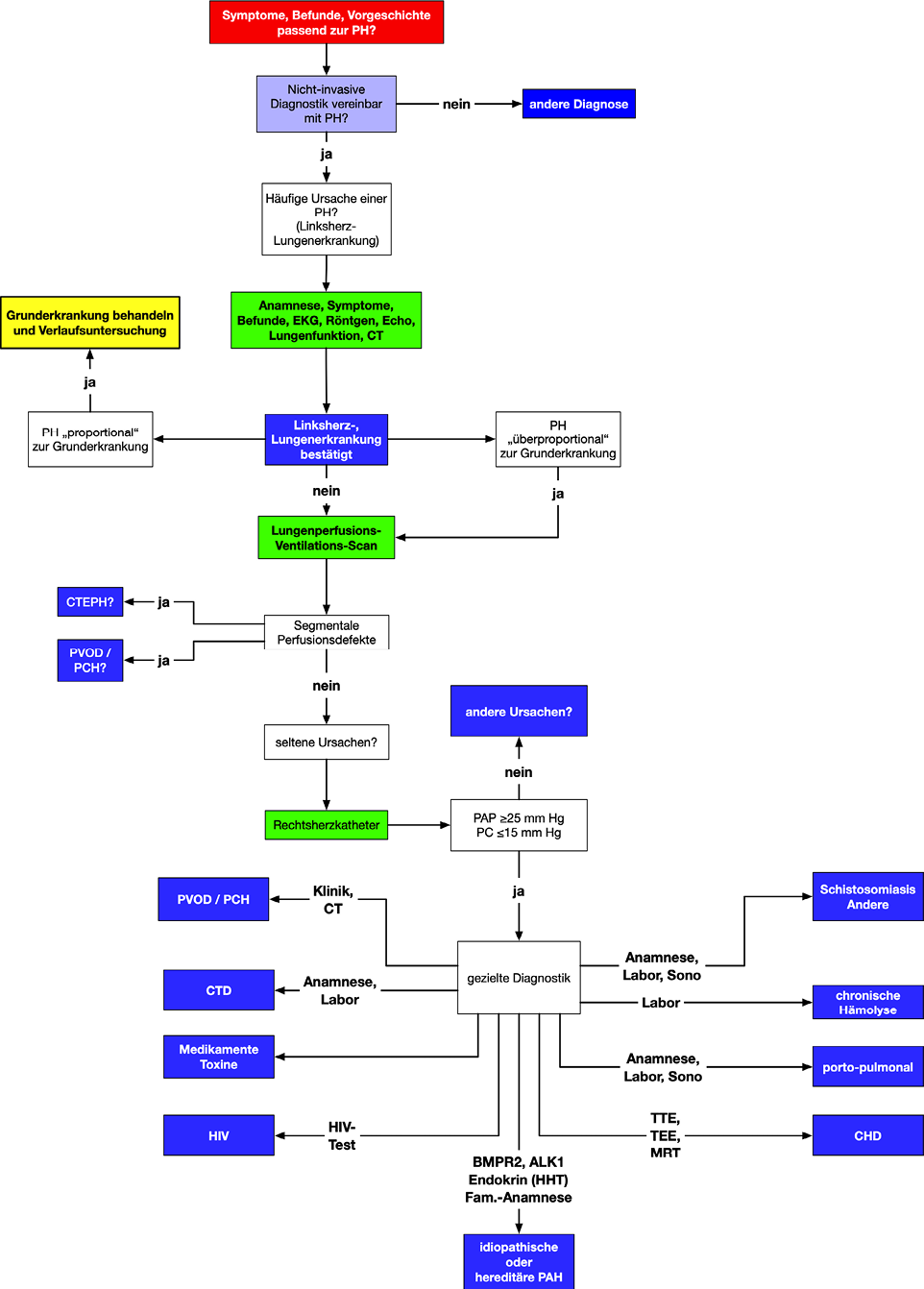
Abb. 12
Diagnostischer Algorithmus bei pulmonaler Hypertonie
Der diagnostische Algorithmus ist in Abb. 12 dargestellt.
Die Strategie zielt darauf ab, zunächst die häufigeren Formen zu identifizieren (Lungen-, Herzerkrankungen), und fokussiert dann auf der Unterscheidung zwischen chronisch thromboembolischen und den verschiedenen anderen Formen der pulmonalen Hypertonie.
Einschätzung des Schweregrads, prognostisch relevante Parameter und Therapieziele
Die Einschätzung des Schweregrads einer pulmonalen Hypertonie erfolgt unter Anwendung komplementärer Methoden, zu denen neben der Anamnese und einer klinischen Untersuchung auch Belastungstests, Laboruntersuchungen, Echokardiographie und Rechtsherzkatheteruntersuchung gehören.
Mit Hilfe dieser Untersuchungen wird die funktionelle Klasse nach der WHO-Nomenklatur (siehe dort) der Erkrankung bestimmt.
Definition des Patientenstatus
Anhand der o.g. Parameter wird der Patientenstatus wie folgt definiert:
Patienten, deren Parameter sich in Tab. 3 ausschließlich oder überwiegend im grünen Bereich bewegen, werden als stabil auf gutem Niveau bezeichnet, solche mit mehreren Parametern im roten Bereich als instabil und gefährdet.
Patienten, die sich zwischen diesen Zonen befinden, werden als stabil auf unbefriedigendem Niveau charakterisiert. In den letzten beiden Fällen sollte eine Eskalation der Therapie erwogen werden. Therapieziel ist der „grüne Bereich“.
Therapie der pulmonal-arteriellen Hypertonie
Die pulmonale Hypertonie ist eine schwerwiegende und progressive Erkrankung. Obwohl eine Heilung bisher nicht möglich ist, haben neue Therapiemöglichkeiten und Behandlungsstrategien der letzten Jahre zu einer Verbesserung der Prognose geführt.
Generell wird eine pulmonale Hypertonie neben der Beachtung allgemeiner Maßnahmen medikamentös behandelt. Neben supportiven Behandlungsmöglichkeiten wie Gerinnungshemmern, Medikamenten zur Entlastung des rechten Herzens und entwässernden Medikamenten stehen Substanzen mit direkter Wirkung auf die Lungengefäße zur Verfügung.
Die Prognose der Erkrankung kann anhand von verschiedenen klinischen, funktionellen, biochemischen, echokardiographischen und hämodynamischen Parametern abgeschätzt werden. Dabei werden die Patienten basierend auf diesen Parametern in 3 Risikokategorien (niedriges, mittleres und hohes Risiko in Bezug auf die 1-Jahres-Mortalität) eingeteilt.
Weil es sich um eine progressive Erkrankung handelt, sollte das Therapieziel das Erreichen bzw. Aufrechterhalten eines geringen Risikostatus sein.
Die Therapie basiert auf 2 Säulen: Der Allgemeinmaßnahmen und der medikamentösen Therapie.
Allgemeinmaßnahmen
Körperliche Betätigung
Körperliche Überanstrengung sollte auf jeden Fall vermieden werden, da sie zu Synkopen oder akutem Rechtsherzversagen führen kann.
Ein unter ärztlicher Kontrolle angeleitetes dosiertes Ausdauertraining kann die körperliche Belastbarkeit und die Lebensqualität allerdings positiv beeinflussen, wenn verhindert wird, daß sich die Patienten körperlich nicht überlasten.
Sauerstoff-Therapie
Bei Hypoxämie ist, auch unter häuslichen Bedingungen die Gabe von Sauerstoff erforderlich. Die Intensität dieser Behandlung muß in Abhängigkeit von einer evtl. Grunderkrankung der Lungen individuell vom Pneumologen definiert werden, weil z.B. in fortgeschrittenen Fällen einer COPD eine zu intensive Sauerstoffgabe zur Ateminsuffizienz führen kann.
Infektionsprävention
Bei Patienten mit pulmonal arterieller Hypertonie wird eine regelmäßige Influenza- und Pneumokokken-Impfung empfohlen.
Familienplanung
Patientinnen mit pulmonaler Hypertonie müssen eine Schwangerschaft im Allgemeinen vermeiden. Eine entsprechende Beratung sowie eine zuverlässige Kontrazeption sind notwendig, wobei auf pharmakokinetische Interaktionen mit den unten genannten spezifischen Medikamenten hingewiesen werden muß, die die Wirksamkeit hormoneller Kontrazeptiva beeinträchtigen können.
Operationen
Wenn möglich sollte eine Regionalanästhesie, wenn möglich, einer Allgemeinanästhesie vorgezogen werden.
Psychosoziale Unterstützung
Psychologische Betreuung wird bei Depressionen und Angststörungen empfohlen.
Reisen
Reisen in Höhen 1500–2000 m sollten vermieden werden, ebenso wie Flug- reisen, sofern nicht sichergestellt ist, daß die O2-Sättigung während des Flugs >90% beträgt.
Ggfs. sollte während des Fluges eine zusätzliche Sauerstoffgabe in Betracht gezogen werden, was zuvor mit der Fluggesellschaft abgesprochen werden muß.
Es empfiehlt sich außerdem, auf Reisen eine schriftliche Information über die Erkrankung mitzuführen und einen lokalen Kontakt zu einer spezialisierten Klinik zu kennen.
Medikamentöse Therapie
Gezielte Medikamente wirken in der Regel über einen der pathogenen Mechanismen, von denen man weiß, daß sie an der Entstehung der pulmonalen Hypertonie beteiligt sind. Die zielgerichteten Medikamente lassen sich kombinieren, so dass alle Wirkmechanismen für die Therapie genutzt werden können.
Endothelin-Rezeptor-Antagonisten
Der Botenstoff Endothelin wird vom Endothel ausgeschüttet und verursacht Gefäßverengung und übermäßiges Wachstum von Zellen in der Gefäßwand. Dies wird über Bindungsstellen, auch Rezeptoren genannt, an die Zellen vermittelt.
Vor diesem Hintergrund wurden die sogenannten Endothelin-Rezeptor-Antagonisten entwickelt. Diese Substanzen blockieren die Bindungsstellen, so dass Endothelin seine Wirkung nicht ausüben kann.
Alle 3 zugelassenen Substanzen (Bosentan, Sitaxentan und Ambrisentan) gelten als potenziell hepatotoxisch und dürfen nur von registrierten Verschreibern verordnet werden. Regelmäßige Kontrollen der Transaminasen (in der Regel in 4-wöchentlichen Abständen) sind erforderlich. Die unten aufgeführten Zieldosierungen betreffen erwachsene Patienten; für Kinder bzw. Patienten <40 kg KG wird auf die jeweiligen Fachinformationen verwiesen.
- Bosentan ist zugelassen für die primäre pulmonal-arterielle Hypertonie, für diejenigen Hypertonieformen, die mit Bindegewebserkrankungen assoziiert sind, sowie pulmonale Hypertonien bei kongenitalen Vitien in den WHO-Funktionsklassen 2 und 3.
Zieldosis: 2-mal 125 mg/Tag p.o.
Für Kinder ist Bosentan ab einem Alter von 2 Jahren zugelassen und auch in einer speziellen Darreichungsform von 4- fach teilbaren 32 mg Tabletten verfügbar.
- Sitaxentan ist in Deutschland nicht mehr zugelassen.
- Ambrisentan ist zugelassen für die primäre pulmonal-arterielle Hypertonie und für diejenigen Hypertonieformen, die mit Bindegewebserkrankungen assoziiert sind und die in den WHO-Funktionsklassen 2 und 3 sind.
Zieldosis: 1-mal 5 mg/Tag oder 1-mal 10 mg/Tag p.o.
- Macitentan ist zugelassen für die primäre pulmonal-arterielle Hypertonie in den WHO-Funktionsklasse 2 - 3
Dosis: 10 mg Macitentan 1mal täglich
- Selexipag ist zugelassen für die primäre pulmonal-arterielle Hypertonie in den WHO-Funktionsklasse 2 - 3.
Dosis: Zu Beginn der Behandlung Initialdosis von 200 ug zweimal täglich im Abstand von etwa 12 Stunden.
Die Dosis kann dann wöchentlich – je nach individueller Verträglichkeit und Bedürftigkeit – bis zu maximal zweimal täglich 1.600 ug titriert werden.
Phosphodiesterase (PDE)-5-Inhibitoren
- Sildenafil (Viagra) ist zugelassen für die primäre pulmonal-arterielle Hypertonie und für diejenigen Hypertonieformen, die mit Bindegewebserkrankungen assoziiert sind und die in den WHO-Funktionsklassen 2 und 3 sind.
Zugelassene Dosis: 3-mal 20 mg/Tag p.o.; diese Dosis wird von der Mehrzahl der Experten als in vielen Fällen nicht ausreichend angesehen, sodass Dosierungen bis zu 3-mal 80 mg/Tag in Deutschland gebräuchlich sind.
- Tadalafil ist zugelassen für die primäre pulmonal-arterielle Hypertonie und für diejenigen Hypertonieformen, die mit Bindegewebserkrankungen assoziiert sind und die in den WHO-Funktionsklassen 2 und 3 sind.
Dosis: 1-mal 40 mg/ Tag p.o.
Stimulatoren der löslichen Guanylatzyklase (sGC-Stimulatoren)
Stickstoffmonoxid (NO) fungiert in Zellen der glatten Muskulatur als Vasodilatator.
Bei Patienten mit pulmonaler arterieller Hypertonie (PAH) ist das Enzym NO-Synthase, das die NO-Produktion bedingt, nur reduziert vorhanden. Als Resultat findet man bei diesen Patienten geringere Konzentrationen an zellulär produziertem NO und daher eine Vasokonstriktion.
NO bindet an das Enzym Guanylatzyklase (sGC) und induziert so die Produktion von zyklischem Guanosinmonophosphat (cGMP). Dieses cGMP aktiviert eine Proteinkinase, die die intrazelluläre Kalziumionenkonzentration reguliert. Die hierdurch bedingte Änderung des Kalziumspiegels in der Zelle verursacht eine Veränderung der Aktin-Myosin-Kontraktion in den glatten Muskelzellen der Gefäßwände und damit eine Blutgefäßerweiterung.
Bei Vorliegen einer PAH ist dieser Weg gestört, da nicht genügend NO zur Verfügung steht oder es ungenügend wirkt.
Der Wirkstoff Riociguat wirkt als direkter sGC-Stimulator unabhängig von NO, indem er die sGC stimuliert und somit unabhängig von NO die Blutgefäße erweitern.
- Riociguat ist zugelassen bei pulmonal-arterieller Hypertonie der WHO-Funktionsklassen 2 - 3.
Dosis: 1 mg-Tablette 3-mal täglich über 2 Woche
alle 2 Wochen bis zu einem Maximum von 2,5 mg 3-mal täglich steigern (maximale Tagesdosis von 7,5 mg)
Prostanoide
Prostacyclin ist eine gefäßerweiternde Substanz, die natürlicherweise im Körper vorkommt. Durch Bindung an die sogenannten IP-Rezeptoren, wodurch als Folge eine Gefäßerweiterung stimuliert wird.
Bei Patienten mit pulmonaler Hypertonie ist die Konzentration von Prostacyclin im Körper erniedrigt. Dies kann die Ursache für eine Verengung der Blutgefäße in der Lunge sein und daher kann therapeutisch verabreichtes Prostazyklin den pulmonalen Blutdruck senken.
- Prostazyklin PGI2(Epoprostenol) ist zugelassen bei primärer pulmonaler Hypertonie, die nicht ausreichend auf eine andere Therapie anspricht, sowie bei sekundärer pulmonaler Hypertonie im Rahmen einer Sklerodermie-Erkrankung in den WHO-Funktionsklassen 3 und 4.
Dosis: Wegen seiner kurzen Halbwertszeit von nur 3 - 4 min muß Epoprostenol durch Perfusoren über einen zentralvenösem Dauerkatheter kontinuierlich infundiert werden.
Entscheidende Nachteile dieses Verfahrens sind technisch bedingte Therapiepausen, Komplikationen seitens des zentralen Venenkatheters, die bei 10 % der Patienten beobachtet wurden, der hohe technisch-logistische Aufwand und die Therapiekosten.
Neben dem Epoprostenol gibt es weitere Medikamente, die eine ähnliche gefäßerweiternde Wirkungsweise wie Prostacyclin haben, sich aber chemisch und pharmakologisch von ihr unterscheiden. Man subsumiert diese Substanzen wegen ihrer auf Prostazyklin-beruhenden Wirkweise als Prostanoide:
IP-Rezeptor-Agonisten
- Selexipag ist zugelassen bei pulmonal-arterieller Hypertonie der WHO-Funktionsklasse 2 - 3.
Dosis: Individuelles Hochtitrieren bis zur höchsten individuell verträglichen Dosis (200 und 1.600 µg 2mal täglich).
Anfangsdosis 2mal täglich 200 µg.
Steigerung üblicherweise wöchentlich in Schritten von 200 µg 2mal täglich (Maximaldosis: 1.600 ug 2mal täglich).
Prostacyclin-Analoga
Iloprost ist ein Prostazyklinderivat mit identischem Wirkspektrum wie Prostazyklin, aber zehnfach längerer Halbwertszeit (ca. 20 bis 30 min) und klinischer Wirkdauer von bis zu 2 Stunden.
Iloprost wird bei inhalativer Anwendung über einen speziellen Vernebler in 6 - 9 Einzeldosen über den Tag verteilt eingenommen werden.
Gegenüber der intravenösen Applikation von Prostazyklin hat inhalatives Iloprost den Vorteil, daß es zu einer geringeren Absenkung des Blutdrucks im Systemkreislauf führt als intravenöses Prostazyklin, sowie daß es eine Abnahme intrapulmonaler Shunts bewirkt, wodurch die arterielle Sauerstoffsättigung ansteigt.
So kommt es im Gegensatz zu intravenös appliziertem Prostazyklin unter inhalativem Iloprost zu einer nur marginalen Absenkung des systemischen Gefäßwiderstandes, während der pulmonalarterielle Mitteldruck stärker gesenkt wird bei gleichzeitig geringerem Anstieg des Herzzeitvolumens.
Während intravenöses Prostazyklin zu einem Abfall des arteriellen Sauerstoffpartialdruckes und der arteriellen Sauerstoffsättigung führt, bleiben diese Parameter nach Inhalation von Iloprost auf hohem Niveau stabil.
Stickstoffmonoxid (NO)
Stickstoffmonoxid (NO) ist ein Signalmolekül in den Zellen der glatten Muskulatur, das über die Aktivierung der löslichen Guanylatzyklase die Bildung des Botenstoffes cGMP auslöst und damit eine Erweiterung der Blutgefäße bewirkt. cGMP wird durch das Enzym PDE-5 wieder abgebaut.
Bei Patienten mit pulmonaler Hypertonie ist die NO-Synthese reduziert. Daher ist eine weitere neue Therapieoption die inhalative Gabe von Stickstoffmonoxid.
Hier verhindern aber bisher ungelöste technische Probleme einen breiten Einsatz.
Kalzium-Antagonisten
Bei entsprechend sensitiven Patienten führen Kalziumantagonisten in hoher Dosierung zu einer Verbesserung der Überlebensraten. Dies betrifft aber nur 20 bis 30 % der Patienten, vornehmlich in den WHO-Funktionsklassen 1 und 2.
Eine kleine Gruppe (<10%) der Patienten mit primärer pulmonal-arterieller Hypertonie spricht auch langfristig auf Kalzium-Antagonisten in hoher Dosierung an. Dies muss zuvor bei einer obligaten Rechtsherzkatheteruntersuchung (Vasoreaktivitätstest) invasiv festgestellt und regelmäßig überprüft werden.
Kalzium-Antagonisten werden ausdrücklich nur bei primärer pulmonal-arterieller Hypertonie (nicht bei anderen Formen der pulmonalen Hypertonie) empfohlen, die die oben beschriebenen Responder-Kriterien erfüllen.
Die Therapie sollte nur in Zentren begonnen werden, die umfangreiche Erfahrungen mit solchen Patienten haben, da eine nicht indizierte Therapie mit Kalzium-Antagonisten fatale Konsequenzen haben kann.
Obgleich die meisten Daten zu Nifedipin und Diltiazem vorliegen, wird in Deutschland mittlerweile an den meisten Zentren überwiegend Amlodipin eingesetzt, weil die Langzeittherapie durch die systemische Blutdrucksenkung unter der hoch dosierten Therapie mit Nifedipin oder Diltiazem limitiert ist.
Die initiale Dosis Amlodipin beträgt 2,5 mg/Tag.
Die Dosis wird unter Beobachtung des Patienten im Laufe einiger Wochen auf die Zieldosis von 10 - 20 mg/d gesteigert.
Patienten, die unter dieser Therapie nicht in die WHO-Funktionsklasse 1 oder 2 übergeführt werden können und deren Hämodynamik sich nicht weitgehend normalisiert, sollten nicht mit Kalzium-Antagonisten, sondern mit den oben aufgeführten spezifischen Substanzen behandelt werden.
Medikamenteninteraktionen
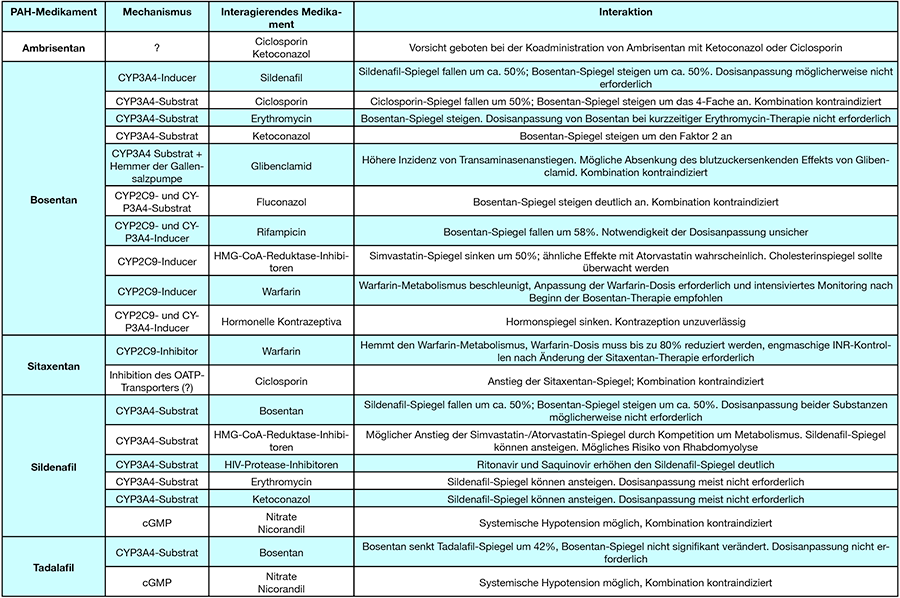
Bei der Behandlung mit den oben genannten spezifischen Medikamenten muss bedacht werden, daß zum Teil klinisch relevante Medikamenteninteraktionen bestehen können.
Die wichtigsten pharmakokinetischen Interaktionen sind in Tab. 5 zusammengestellt.
Tab. 5: INR„international normalized ratio“, cGMP zyklisches Guanosin-Monophosphat, OATP„organic anion transporter proteins“.
Kombinationstherapie
Da die pulmonale Hypertonie nicht heilbar ist und die Therapieziele bei der Mehrzahl der Patienten mit Monotherapie nicht bzw. nicht dauerhaft zu erreichen sind kann man die Medikamente auch kombinieren.
Die bislang dazu vorliegenden Daten weisen auf eine hohe Sicherheit der Kombinationstherapie hin und liefern Hinweise für verbesserte Behandlungsresultate. Allerdings ist die Datenlage zur Kombinationstherapie noch unzureichend.
Am gebräuchlichsten ist die Kombination eines Endothelin-Rezeptor-Antagonisten mit einem PDE-5- Inhibitor, aber konkrete Empfehlungen für den Einzelfall können aufgrund mangelnder Daten nicht gegeben werden.
Therapiealgorithmus
Ziel aller Behandlungen ist eine O2-Sättigung ≥92%.
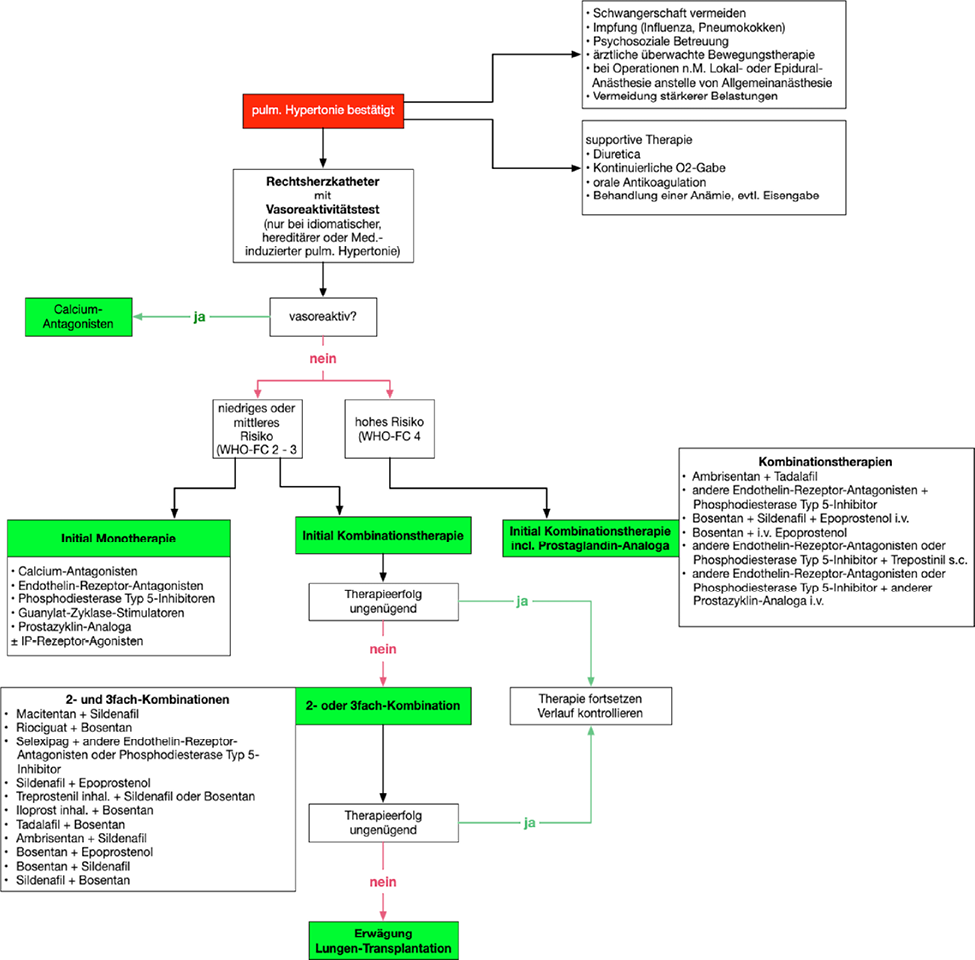
Der aktuelle Therapiealgorithmus für Patienten mit primärer pulmonal-arterieller Hypertonie ist in Abb. 13 dargestellt. Diese Empfehlungen gelten nicht für andere Formen der pulmonalen Hypertonie.
Abb. 13
Therapiealgorithmus für Patienten mit pulmonal-arterieller Hypertonie (gilt nur für Patienten der Gruppe 1).
aus: Diagnostik und Therapie der pulmonalen Hypertonie, Europäische Leitlinien 2015
Für andere Formen der pulmonalen Hypertonie gelten unter Umständen spezielle Hinweise, auf die weiter unten eingegangen wird.
Supportive Therapie
Orale Antikoagulantien
Bei pulmonaler Hypertonie liegt (in Abhängigkeit von ihrer Ätiologie) eine erhöhte Neigung zur Blutgerinnselbildung in den Lungengefäßen vor, sodass im Einzelfall erwogen werden muss, ob die Einnahme von gerinnungshemmenden Medikamenten erforderlich ist.
Trotz unzureichender Datenlage wird empfohlen, Patienten mit primärer pulmonal-arterieller Hypertonie mit einem oralen Antikoagulans zu behandeln, sofern keine Kontraindikationen vorliegen.
Dies gilt auch für die orale Antikoagulation bei der hereditären pulmonal-arteriellen Hypertonie und bei pulmonaler Hypertonie nach Einnahme von Appetitzüglern.
Hingegen besteht bei Patienten mit einer auf einer Lungenkrankheit basierenden pulmonalen Hypertonie nur eine IIb-Empfehlung (Nutzen übersteigt das Risiko oder gleicht dem Risiko).
Bei Patienten mit chronisch thromboembolisch bedingter pulmonaler Hypertonie ist der lebensverlängernde Wert der Antikoagulation erwiesen.
Die Ziel-INR beträgt 2,0–3,0.
Diuretika
Bei einer Rechtsherzinsuffizienz, die in fortgeschrittenen Stadien der pulmonalen Hypertonie auftritt kommt es zu peripheren Ödemen und/oder auch zur (schmerzhaften) Hepatomegalie.
Hier werden Diuretika nach klinischer Indikation eingesetzt.
Es gibt keine Daten zur Über- bzw. Unterlegenheit einzelner Substanzen.
Sauerstoff
Eine Behandlung mit Sauerstoff führt in der Regel zwar nicht zu einer relevanten Drucksenkung im Lungenkreislauf, kann jedoch das Allgemeinbefinden bessern. Die Gabe von Sauerstoff sollte in Betracht gezogen werden, wenn die Sauerstoffsättigung <90% bzw. der Sauerstoff-Partialdruck im Blut dauerhaft <60 mm Hg beträgt.
Sie orientiert sich weitgehend an den Empfehlungen für Patienten mit COPD. Für die primäre pulmonal-arterielle Hypertonie liegen keine Daten vor, sodaß die Entscheidung zur O2-Therapie individuell getroffen wird.
Behandlung von Arrhythmien
Während ventrikuläre Tachykardien bei Patienten mit pulmonaler Hypertonie nur selten auftreten, werden supraventrikuläre Tachykardien, v. a. Vorhofflattern und -flimmern, häufig beobachtet.
Solche Arrhythmien führen auch bei normaler Kammerfrequenz unbehandelt meist zu einer deutlichen klinischen Verschlechterung mit zunehmender Rechtsherzinsuffizienz. Diese Patienten profitieren von einer aggressiven Therapie mit dem Ziel, den Sinusrhythmus wiederherzustellen. Obgleich die Datenlage zur Behandlung dieser Rhythmusstörungen spärlich ist, sollte, wenn möglich, eine Wiederherstellung bzw. der Erhalt des Sinusrhythmus angestrebt werden.
Eisensubstitution
Eisenmangel ist ein häufiges Phänomen bei pulmonal arterieller Hypertonie, daher sollte der Eisenwert regelmäßig überprüft und bei Eisenmangel eine Substitutionstherapie in Betracht gezogen werden.
Glykoside
Bei Funktionseinschränkungen des rechten Herzens können Glykoside oder andere kardiovaskuläre Medikamente eingesetzt werden, vor allem bei bestimmten Arrhythmien, wie z.B. Vorhofflimmern.
Atrioseptostomie
Eine frühzeitige Atrioseptostomie verbessert möglicherweise die Prognose von Patienten mit primär pulmonal-arterieller Hypertonie, wobei es keine Daten dazu gibt, ob dies auch für Patienten gilt, die mit spezifischen Medikamenten behandelt werden.
Die Atrioseptostomie ist bei medikamentös behandelten Patienten meist nicht indiziert und kann nicht generell empfohlen werden. Als Notfallmaßnahme bei dekompensiertem Rechtsherzversagen sollte sie nicht eingesetzt werden.
Derzeit wird dieses Verfahren in Deutschland nur sehr selten angewandt.
Lungentransplantation
Die Einzel-, Doppel-Lungen- bzw. Herz-Lungen-Transplantation stellt weiterhin ein Therapieverfahren für geeignete Patienten dar, bei denen unter optimierter konservativer Therapie kein ausreichendes Behandlungsergebnis erzielt werden kann. Potenzielle Kandidaten für eine Transplantation sollten frühzeitig an einem geeigneten Zentrum vorgestellt werden.
Dies gilt sowohl für die Patienten mit primär pulmonal-arterieller als auch diejenigen mit sekundärer pulmonaler Hypertonie. Voraussetzung ist, daß die Grunderkrankung ausbehandelt ist beziehungsweise wenn Risikofaktoren soweit als möglich ausgeschaltet sind (zum Beispiel Absetzen von Appetitzüglern).
Spezielle Populationen mit pulmonaler Hypertonie
Pädiatrische Patienten
Diese Patienten sollten in Expertenzentren behandelt werden. Die Komplexität der Diagnostik und Therapie unterschiedlicher Formen der pulmonal-arteriellen Hypertonie bei diesen Patienten und unterschiedliche Altersgruppen macht eine ausführliche Darstellung an dieser Stelle unmöglich, sodaß auf die europäischen Leitlinien verwiesen wird.
Pulmonale Hypertonie bei angeborenen Herzfehlern
Auch für Details zu dieser Patientengruppe wird auf die europäischen Leitlinien verwiesen.
Pulmonale Hypertonie bei Bindegewebserkrankungen
Am häufigsten tritt eine pulmonale Hypertonie bei Patienten mit systemischer Sklerose auf (5– 15%), gefolgt von Patienten mit Mischkollagenosen (5–10%) und systemischem Lupus erythematodes (SLE; 2– 5%).
Ein jährliches echokardiographisches Screening auch asymptomatischer Sklerodermiepatienten auf eine pulmonale Hypertonie wird daher empfohlen.
Beim Auftreten typischer Symptome sollte unverzüglich eine entsprechende Abklärung erfolgen, wobei die Diagnostik und Therapie bei diesen Patienten weitgehend dem oben Beschriebenen entspricht.
Der Stellenwert einer zusätzlichen immunsuppressiven bzw. immunmodulatorischen Therapie hängt von der Grunderkrankung ab:
Bei Sklerodermie-assoziierter pulmonaler Hypertonie gilt dieser Ansatz als wirkungslos und wird nicht empfohlen, während bei systemischem Lupus erythematodes gute Erfolge mit immunsuppressiver Behandlung beschrieben wurden.
Patienten mit Mischkollagenose-assoziierter pulmonaler Hypertonie verhalten sich meist wie Sklerodermiepatienten, können aber in Einzelfällen auch von immunsuppressiver Therapie profitieren.
Portopulmonale Hypertonie
Sie ist definiert als das Auftreten einer pulmonalen Hypertonie bei Patienten mit portaler Hypertension. Die Inzidenz bei Patienten mit Leberzirrhose scheint bei 0,5–1% zu liegen. Die Lebererkrankung kann dabei asymptomatisch sein, sodaß bei Patienten mit ätiologisch unklarer pulmonaler Hypertonie gezielt, d. h. sonographisch, auch nach einer portalen Hypertension gesucht werden sollte.
Auf der anderen Seite muss bei Patienten mit Leberzirrhose und Belastungsdyspnoe auch an eine pulmonale Hypertonie gedacht werden.
Bei Patienten, die zur Lebertransplantation evaluiert werden, sollte grundsätzlich eine Echokardiographie zum Ausschluß einer Rechtsherzbelastung durchgeführt werden, da die Prognose nach Transplantation bei Vorliegen einer pulmonalen Hypertonie äußerst schlecht ist.
Kontrollierte Daten zur Therapie dieser Form der pulmonalen Hypertonie liegen praktisch nicht vor. Bei Patienten mit Child-A-Zirrhose, d. h. nur leicht eingeschränkter Leberfunktion, sind positive Erfahrungen mit Bosentan und Sildenafil publiziert. Bei fortgeschrittener Lebererkrankung sind Endothelin-Rezeptor-Antagonisten kontraindiziert, sodaß hier vornehmlich PDE-5- Hemmer und/oder Prostanoide eingesetzt werden.
In seltenen Einzelfällen kommt bei therapierefraktärem Krankheitsbild eine kombinierte Lungen-Leber-Transplantation in Betracht.
Pulmonale Hypertonie bei HIV-Infektion
Etwa 0,5% aller HIV-infizierten Patienten entwickeln eine pulmonale Hypertonie. Daher gehört eine Untersuchung auf das Vorliegen einer HIV- Infektion gehört zur Standarddiagnostik bei Patienten mit ätiologisch unklarer pulmonaler Hypertonie.
Auch hier orientiert sich die Therapie weitgehend an den oben genannten Empfehlungen. Positive Erfahrungen wurden mit Bosentan und Sildenafil beschrieben.
In einigen, aber nicht in allen Fällen konnte auch der Einsatz einer antiretroviralen Therapie den Verlauf der pulmonalen Hypertonie günstig beeinflussen.
Zu beachten sind potenzielle Interaktionen zwischen den spezifischen Medikamenten der pulmonalen Hypertonie und der antiretroviralen Therapie. Dies gilt v. a. für die Kombination von Sildenafil mit Protease-Inhibitoren.
Pulmonale venookklusive Erkrankung
Diese seltene Form kann gelegentlich durch typischen Veränderungen im CT erkannt werden, die allerdings bei der Mehrzahl der Patienten nicht nachweisbar sind. Somit ist eines der wichtigsten Kriterien für das mögliche Vorliegen einer venookklusiven Erkrankung das unzureichende Ansprechen bzw. eine klinische Verschlechterung unter einer spezifischen Therapie.
Die pulmonale venookklusive Erkrankung gilt nach wie vor als medikamentös nicht behandelbar und hat eine ausgesprochen schlechte Prognose, sodaß Patienten schon bei entsprechender Verdachtsdiagnose unverzüglich an einem Transplantationszentrum vorgestellt werden sollten.
Pulmonale Hypertonie bei Linksherzerkrankungen
Es handelt sich um eine der häufigsten Formen der pulmonalen Hypertonie, deren Auftreten die Prognose von Patienten mit Linksherzinsuffizienz verschlechtert.
Bei bekannter Linksherzinsuffizienz mit eingeschränkter systolischer Funktion des linken Ventrikels ist die Diagnose meistens nicht schwierig. Bei überwiegend oder sogar rein diastolischer Dysfunktion des linken Ventrikels kann die Abgrenzung zu einer primär pulmonal-arteriellen Hypertonie schwierig sein.
Typische echokardiographische Zeichen einer diastolischen LV-Funktionsstörung sind ein vergrößerter linker Vorhof, charakteristische Flussprofile über der Mitralklappe und den Lungenvenen, sowie eine linksventrikuläre Hypertrophie. Häufig haben diese Patienten ein permanentes Vorhofflimmern. Auch der BNP-Wert ist zumeist z.T. deutlich erhöht.
Die nachfolgende Liste enthält typische Faktoren, die mit einer diastolischen LV-Funktionsstörung assoziiert sein können:
- Alter >65 Jahre
- Arterieller Hypertonus (auch anamnestisch)
- Adipositas, metabolisches Syndrom
- Koronare Herzerkrankung
- Diabetes mellitus
- Vorhofflimmern
In der Regel ist eine Rechtsherzkatheteruntersuchung erforderlich, um zuverlässig zwischen einer pulmonalen Hypertonie bei Linksherzinsuffizienz und einer primären pulmonal-arteriellen Hypertonie zu unterscheiden:
Ein erhöhter PC-Druck (>15 mm Hg) belegt das Vorliegen einer LV-Funktionsstörung, ein normaler PC schließt diese aber nicht aus, insbesondere bei diuretisch vorbehandelten Patienten. In Zweifelsfällen wird eine Volumenbelastung (z.B. 500 ml NaCl 0,9% über 5 – 10 min) oder eine Belastungsuntersuchung empfohlen, um eine LV-Dysfunktion nachzuweisen bzw. auszuschließen. Diese Testverfahren sind allerdings bislang nicht hinreichend standardisiert.
Ein transpulmonaler Gradient >12 mm Hg spricht für eine pulmonal-arterielle Komponente.
Bislang gibt es keine gezielte Therapie der pulmonalen Hypertonie bei Linksherzerkrankungen. Bei manifester Herzinsuffizienz wird primär eine leitliniengerechte Therapie der zugrunde liegenden Linksherzerkrankung empfohlen. Keine der bei Linksherzinsuffizienz empfohlenen Substanzen ist bei begleitender pulmonaler Hypertonie kontraindiziert.
Kontrollierte Studien mit Prostacyclin-Analoga und Bosentan zeigten keinen Nutzen dieser Substanzen bei Linksherzinsuffizienz, im Falle von intravenösem Prostacyclin wurde sogar eine höhere Sterblichkeit der Behandlungsgruppe festgestellt. Für Sildenafil gibt es einzelne experimentelle und unkontrollierte klinische Daten, die jedoch keine generelle Empfehlung zulassen, zumal der Effekt auf die Langzeitprognose dieser Patienten noch nicht untersucht wurde.
Pulmonale Hypertonie bei chronischen Lungenerkrankungen
Sowohl bei Patienten mit chronisch obstruktiven Lungenerkrankungen (COPD) als auch bei solchen mit interstitiellen Lungenerkrankungen tritt eine pulmonale Hypertonie häufig auf, in fortgeschrittenen Fällen teilweise in >50% der Fälle.
In der Regel verläuft die Hypertonie mild und zeigt andere Charakteristika als die primäre pulmonal-arterielle Hypertonie, d. h., die Pulmonalisdrücke sind weniger stark erhöht (PA-Mitteldruck selten >35 mm Hg), das Herzzeitvolumen bleibt meist normal und der pulmonal-vaskuläre Widerstand ist somit meist nur leicht erhöht (Tab. 6).

Links: Tab. 6
Hämodynamische Klassifizierung der pulmonalen Hypertonie bei Lungenerkrankungen
Abkürzungen: CI = cardiac index; HZV = Herzzeitvolumen; COPD = Chronisch obstructive Lungenerkrankung; CPFE = Kombinierte Lungenfibrose und -emphysem; IPF = Idiopathische Lungenfibrose; PAP = Pulmonalarteriendruck; PAPm = Mittlerer Pulmonalarteriendruck; PH = Pulmonale Hypertonie
aus: Diagnostik und Therapie der pulmonalen Hypertonie, Europäische Leitlinien 2015
Dennoch gibt es zahlreiche Hinweise dafür, daß auch leichte Formen einer pulmonalen Hypertonie bei Patienten mit chronischen Lungenerkrankungen von prognostischer Bedeutung sind.
Bislang gibt es weder für die COPD noch für andere mit Lungenerkrankungen assoziierten pulmonalen Hypertonien belastbare Daten, die den Einsatz der spezifischer Medikamente generell rechtfertigen würden. Für Bosentan wurde bei Patienten mit COPD und milder pulmonaler Hypertonie eine Verschlechterung der Oxygenierung festgestellt, die zudem mit einer Verschlechterung der Lebensqualität einherging.
Zum gegenwärtigen Zeitpunkt beschränkt sich die Therapie der pulmonalen Hypertonie bei diesen Patienten auf konventionelle Maßnahmen sowie die Gabe von Sauerstoff entsprechend der jeweiligen Empfehlungen.
Patienten mit eher milder Ausprägung der Lungenerkrankung, aber unverhältnismäßig schwerer pulmonaler Druckerhöhung zeigen gelegentlich die klinischen Charakteristika von Patienten mit primärer pulmonal-arterieller Hypertonie und könnten unter Umständen von einer gezielten Therapie profitieren. Für diese Frage sollte ein spezialisiertes Zentrum kontaktiert werden. In diesen Fällen ist darauf zu achten, daß die Diagnose durch eine Rechtsherzkatheteruntersuchung bestätigt wird, da die Zuverlässigkeit der Echokardiographie besonders bei Patienten mit COPD eingeschränkt ist.
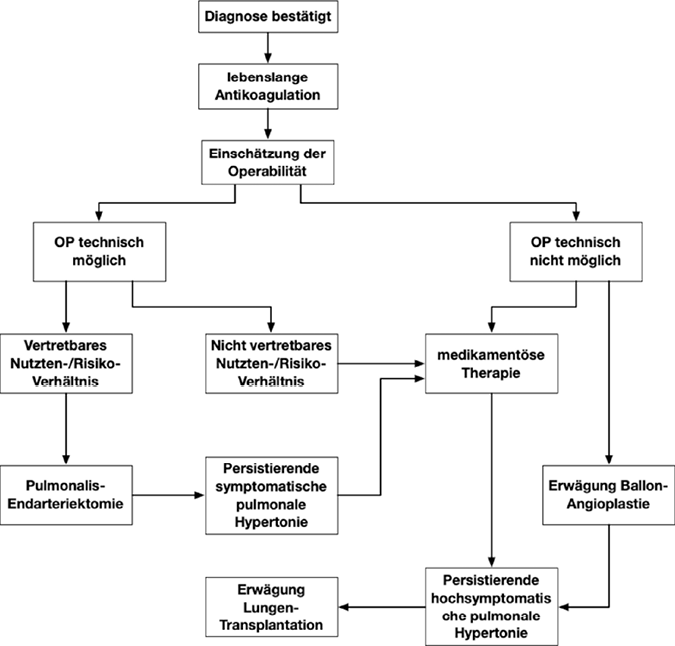
Chronisch thromboembolische pulmonale Hypertonie
Sie ist eine der häufigsten Formen der schweren pulmonalen Hypertonie.
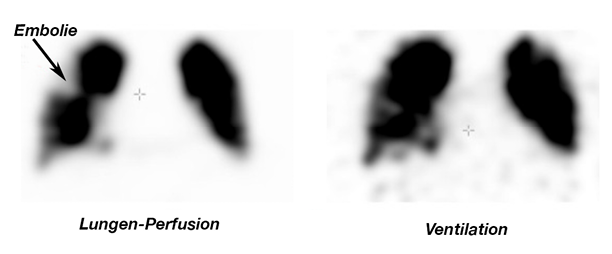
Im Rahmen der Erstdiagnostik einer neu diagnostizierten pulmonalen Hypertonie kommt dem Nachweis bzw. Ausschluss einer thromboembolischen Genese besondere Bedeutung zu. Daher wird in diesen Fällen grundsätzlich eine Ventilations-Perfusions-Szintigraphie empfohlen, da eine normale Lungenperfusion eine chronische Thromboembolien mit hoher Wahrscheinlichkeit ausschließt (Abb. 14).

Abb. 14
Diagnostischer Algorithmus bei Verdacht auf chronisch thromboembolische pulmonale Hypertonie (CTEPH)
aus: Diagnostik und Therapie der pulmonalen Hypertonie, Europäische Leitlinien 2015

.png)